Functional Changes in Metabolic and Malignancy Associated Bacterial Genes in the Post Transplant Intestinal Microbiome
1Multi-Organ Transplant, UHN, Toronto, ON, Canada, 2University of Toronto, Toronto, ON, Canada, 3UHN, Toronto, ON, Canada
Meeting: 2020 American Transplant Congress
Abstract number: 157
Keywords: Hyperglycemia, Liver metabolism, Malignancy, Metabolic disease
Session Information
Session Name: Liver: Immunosuppression and Rejection
Session Type: Oral Abstract Session
Date: Saturday, May 30, 2020
Session Time: 3:15pm-4:45pm
 Presentation Time: 4:27pm-4:39pm
Presentation Time: 4:27pm-4:39pm
Location: Virtual
*Purpose: Long-term survival of liver transplant (LT) recipients is affected by metabolic and malignant complications. The aim of our study was to examine changes in the intestinal microbiome (IM) post-LT and its functions post-transplant, particularly focusing on metabolism- and malignancy-related bacterial genes using inferred functional metagenomics.
*Methods: Stool samples were obtained from the pre-LT baseline and 3- and 6-months post-LT patients. 16S rRNA sequencing (V4 region) was performed on 74 samples from 30 LT recipients. Relative abundances of the community members were determined using rarefied data and summarized as operational taxonomic units (OTUs). Functional analysis of the bacterial genes was determined using PICRUSt and Piphillin tools. Fecal microbiota transplant (FMT) was performed from immunosuppressed rats to germ-free (GF) mice.
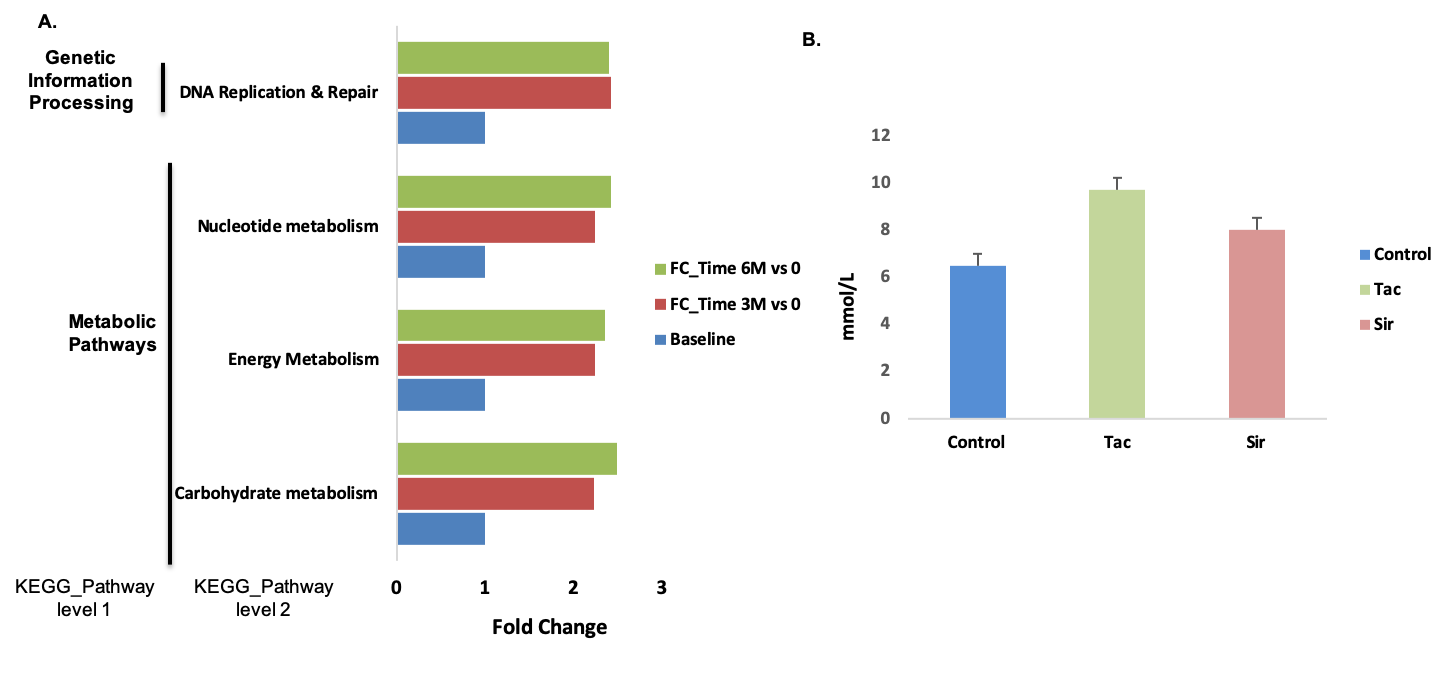
*Results: All patients were on calcineurin inhibitors as immunosuppression. The principal underlying etiologies of liver disease were hepatitis C (30%) and alcoholic cirrhosis (30%). The phylum Firmicutes, in particular, Ruminococcaceae and Lachnospiraceae (family), were significantly abundant at 3- and 6-months, respectively than pre-LT. Conversely, the relative abundance of the family Veillonellaceae was enriched in pre-LT. The functional analysis revealed that genes related to carbohydrate metabolism and DNA replication & repair pathways were significantly increased in overtime after LT. We validated these inferred functional metagenomic profiles using metagenomic sequencing data from rats exposed to tacrolimus and sirolimus. The immunosuppressive drugs induced hyperglycemia in rats. Fecal microbiota transplant (FMT) into germ-free mice induced hyperglycemia, suggesting a contributing causal role of IM.
*Conclusions: Using inferred functional metagenomics, we discovered longitudinal changes in the IM, with the increasing presence of bacterial genes involved in carbohydrate metabolism and DNA replication & repair over time post-transplant. Our results were further strengthened through validation with metagenomics sequencing data from animals exposed to immunosuppressants and with FMT into germ-free mice. LT recipients also had increasing proportions of Firmicutes phyla particularly Lachnospiraceae associated with susceptibility to post-transplant diabetes. These results suggest a potential contribution of the IM to long-term metabolic and malignant complications post-transplant.
To cite this abstract in AMA style:
Bhat M, Sivaraj S, Pasini E, Copeland J, Allard J, Humar A. Functional Changes in Metabolic and Malignancy Associated Bacterial Genes in the Post Transplant Intestinal Microbiome [abstract]. Am J Transplant. 2020; 20 (suppl 3). https://atcmeetingabstracts.com/abstract/functional-changes-in-metabolic-and-malignancy-associated-bacterial-genes-in-the-post-transplant-intestinal-microbiome/. Accessed March 30, 2026.« Back to 2020 American Transplant Congress