Calcineurin Inhibitor (CNI) Nephrotoxicity Is Mediated by Defective Amp-Activated Protein Kinase (AMPK) Activation.
Medicine, UAB, Birmingham, AL.
Meeting: 2016 American Transplant Congress
Abstract number: D75
Keywords: Calcineurin, Immunosuppression, Nephrotoxicity, Oxidant stress
Session Information
Session Name: Poster Session D: Chimerism/Stem Cells, Cellular/Islet Transplantation, Innate Immunity, Chronic Rejection
Session Type: Poster Session
Date: Tuesday, June 14, 2016
Session Time: 6:00pm-7:00pm
 Presentation Time: 6:00pm-7:00pm
Presentation Time: 6:00pm-7:00pm
Location: Halls C&D
Background: CNIs are potent immunosuppressant therapies, but associated with nephrotoxicity manifested by interstitial fibrosis and progressive graft dysfunction. AMPK is a major sensor and regulator of bioenergetics and redox homeostasis, and an essential regulator of mitophagy. Defective AMPK activation is associated with kidney injury in other disease models and augmenting AMPK ameliorates kidney injury. Herein, we investigate the potential link between CNI nephrotoxicity and defective renal bioenergetics.
Methods: In addition to proximal epithelial cells (PTECs), we studied a mouse model of CsA nephrotoxicity where animals were placed on 0.01% NaCl for 4 weeks, followed by CsA 30 mg/kg IP daily.
Results: Exposure of PTECs to CsA results in a potent oxidative stress response, generating cellular and mitochondrial ROS, detected by H2DCFDA-fluorescence and MitoSox superoxide specific probe, respectively (Fig 1A-B). Confocal microscopy reveals substantial alteration of the mitochondrial network, indicative of fission (Fig 1C). Oxidative stress was accompanied by significant alterations in mitochondrial structure, including diminished Electron Transport Chain complex I (3.1±0.1-fold; p<0.05) and complex IV (2.2±0.3-fold; p<0.05) compared to vehicle-treated cells. Further, CsA treatment results in submaximal activation of AMPK as detected by T172-AMPK phosphorylation (2.3±0.4-fold compared to veh; p<0.05), that is significantly augmented by the AMPK activator AICAR (4.9±0.7-fold compared to veh; p<0.05). In vivo, loss of AMPK activity is seen in both cortex and medulla of nephrotoxic mice as early as 2 weeks of CsA treatment (FIg 2). By 8 weeks, active-AMPK remains depressed and is associated with induction of myofibroblast activation as detected by induction of αSMA staining.
Conclusions: CsA treatment alters mitochondrial redox homeostasis with inadequate AMPK activation, associated myofibroblast activation, inciting a pro-fibrogenic renal milieu. Collectively, these results suggest that augmentation of the AMPK-mitochondrial pathway may ameliorate CNI-mediated renal injury. The available clinical AMPK activator, metformin, provides an exciting possibility for studies in man.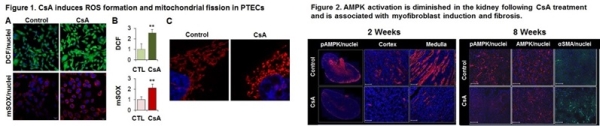
CITATION INFORMATION: Zmijewska A, Chen J, Zmijewski J, Mannon R. Calcineurin Inhibitor (CNI) Nephrotoxicity Is Mediated by Defective Amp-Activated Protein Kinase (AMPK) Activation. Am J Transplant. 2016;16 (suppl 3).
To cite this abstract in AMA style:
Zmijewska A, Chen J, Zmijewski J, Mannon R. Calcineurin Inhibitor (CNI) Nephrotoxicity Is Mediated by Defective Amp-Activated Protein Kinase (AMPK) Activation. [abstract]. Am J Transplant. 2016; 16 (suppl 3). https://atcmeetingabstracts.com/abstract/calcineurin-inhibitor-cni-nephrotoxicity-is-mediated-by-defective-amp-activated-protein-kinase-ampk-activation/. Accessed May 16, 2026.« Back to 2016 American Transplant Congress